Table of Contents
The medical device industry plays a critical role in advancing healthcare globally, and ensuring patient safety and product efficacy is of utmost importance. In the European Union (EU), medical device manufacturers must comply with specific regulations to ensure their products meet the highest standards. The two key regulations governing medical devices in the EU are the EU Medical Device Regulation (EU MDR) and the EU In Vitro Diagnostic Regulation (EU IVDR). In this article, we will delve into the compliance requirements outlined by these regulations and their implications for manufacturers.
What is EU MDR Medical Device Regulation:
The Medical Device Regulation (EU MDR), effective from May 26, 2021, represents a significant overhaul of the previous directives. This regulation was established to replace the Medical Device Directive (93/42/EEC) and the Directive for Active Implantable Medical Devices (90/385/EEC). The MDR introduces enhanced requirements for medical device manufacturers, focusing on rigorous clinical evaluation and market surveillance to improve device quality and safety. The broadened scope of the regulation covers more types of devices, increasing the accountability of manufacturers and other stakeholders in the supply chain.
Key Changes: One of the most notable changes under the MDR is the requirement for a comprehensive quality management system and the mandatory involvement of a European-authorized representative for non-EU manufacturers.
Impact: The MDR aims to reduce the risks associated with medical devices on the market by enhancing transparency and traceability throughout the device's lifecycle.
The broad scope of EU MDR Section 10.4.1:
Section 10.4.1 of the EU Medical Device Regulation (EU MDR) plays a crucial role in ensuring the safety and quality of medical devices. This section requires manufacturers to identify certain substances in their products. Specifically, manufacturers must identify carcinogens, mutagens, reproductive toxins (CMRs), and endocrine-disrupting chemicals (EDCs) that are present in invasive devices or materials that come into contact with fluids or gases. These substances must be identified if they are present in concentrations exceeding 0.1% w/w.
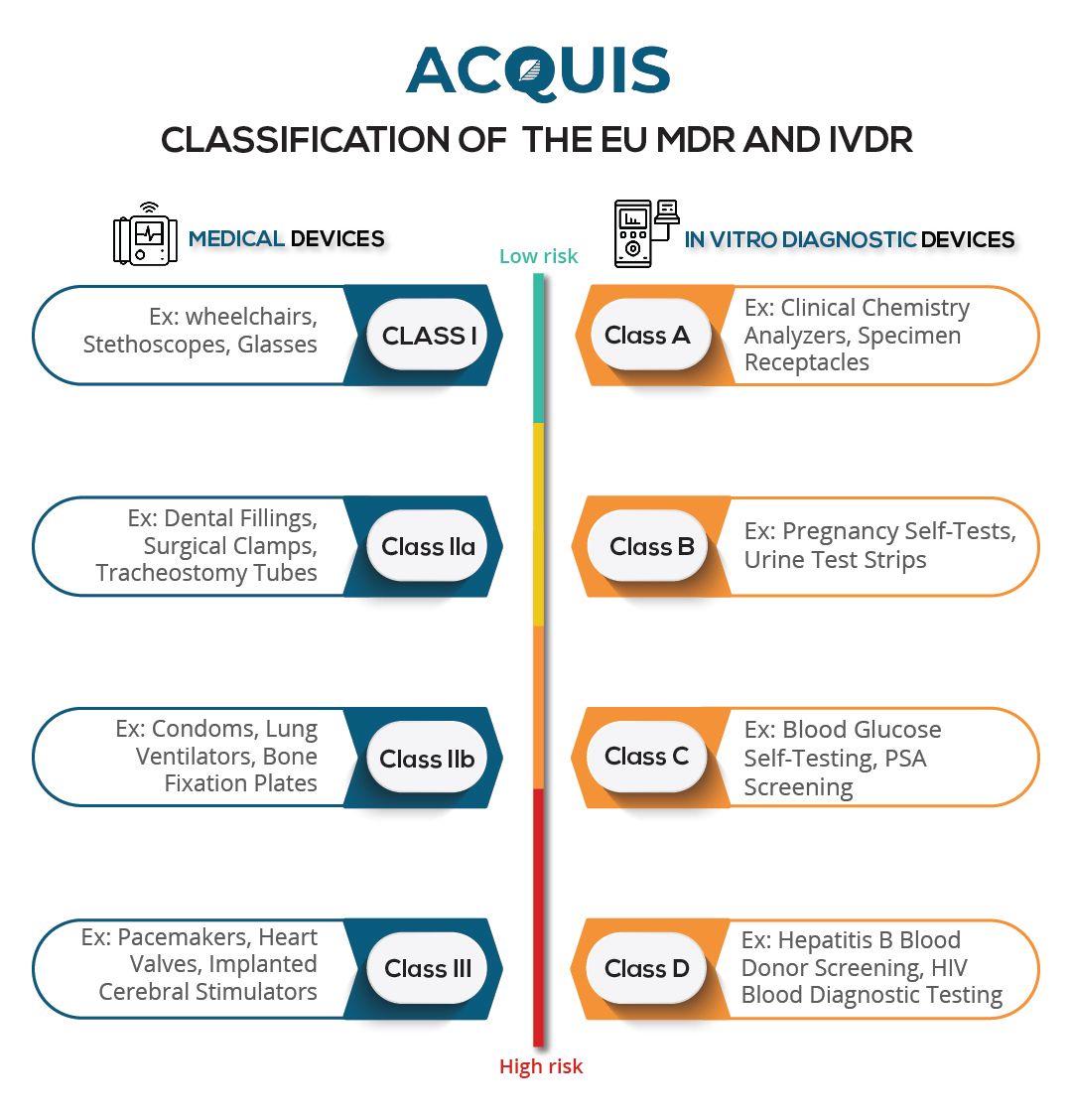
Under the EU MDR, medical device manufacturers must adhere to several key compliance requirements. First and foremost, manufacturers are required to classify their devices based on risk and assign them to one of four classes: Class I, Class IIa, Class IIb, or Class III. This classification determines the level of scrutiny and conformity assessment procedures required.
The classification process follows 22 rules outlined in EU MDR Annex VIII. Rules 19–22 are new to the EU MDR, while rules 1–18 have been carried over from the previous MDD. By considering the three criteria specified in Section 10.4.1, in conjunction with these classification rules, companies can determine the impact of the EU MDR on their products.
Manufacturers must also compile and maintain comprehensive Technical Documentation, which includes detailed information on the design, manufacturing, and performance of the device. This documentation should demonstrate compliance with the general safety and performance requirements outlined in the regulation. It should also include a Clinical Evaluation Report (CER) based on clinical data collected to support the device's safety and performance claims.
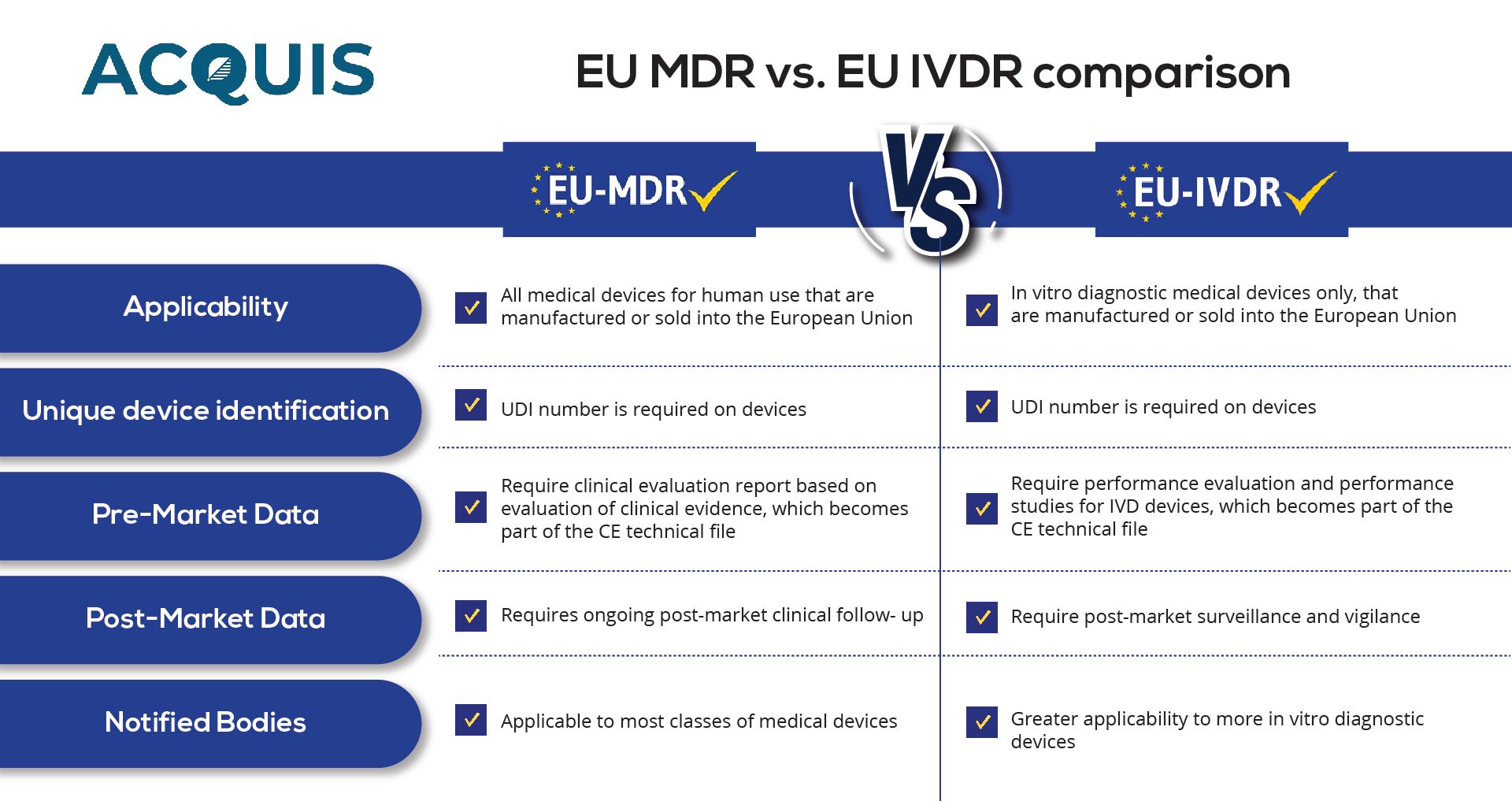
Another important aspect of EU MDR compliance is the requirement for a Notified Body. Notified Bodies are independent organizations designated by EU member states to assess the conformity of medical devices. Manufacturers must obtain a valid certificate from a Notified Body to demonstrate compliance with the regulation. The certification process involves a thorough assessment of the Technical Documentation, Quality Management System, and, in some cases, a review of clinical evidence.
What is EU IVDR In Vitro Diagnostic Regulation
The In Vitro Diagnostic Regulation (EU IVDR) came into effect on May 26, 2022, replacing the older EU Directive on In Vitro Diagnostic Medical Devices (98/79/EC). This regulation sets forth new standards for the approval and surveillance of IVDs, which are tools used to perform tests on samples from the human body. The IVDR introduces a new classification system based on the risk associated with the device, requiring more stringent documentation and compliance for higher-risk classes
Under the EU IVDR, IVD manufacturers face stringent compliance requirements. One significant change is the introduction of a risk-based classification system for IVD devices.
The regulation includes four classes: Class A, Class B, Class C, and Class D. Higher-risk devices will require involvement from a Notified Body to assess conformity.
To comply with the EU IVDR, manufacturers must establish a Quality Management System (QMS) and conduct comprehensive Technical Documentation. The Technical Documentation must include detailed information on the device's design, intended purpose, analytical and clinical performance, as well as an evaluation of any claims made. Clinical evidence, such as performance evaluation studies or clinical performance studies, must support the device's claims.
Like the EU MDR, the EU IVDR also requires involvement with a Notified Body. Manufacturers must obtain a valid certificate from a Notified Body to demonstrate compliance with the regulation. Notified Bodies will review the Technical Documentation and assess the conformity of the device with the regulation's requirements.
Transitional Provisions for Medical Devices and IVDs
The implementation of the MDR and IVDR has introduced transitional provisions that allow certain medical devices and IVDs, referred to as “legacy devices,” to continue being marketed under the previous directives for a limited time. These provisions ensure that manufacturers have sufficient time to meet the new regulatory requirements and obtain the necessary CE certification.
Legacy Devices and Extended Deadlines
Legacy devices are those that were certified under the old directives before the new regulations were enforced. To prevent market disruptions and risks to patient safety, the deadlines for compliance have been extended for both the MDR and IVDR. The IVDR was extended in January 2022, while the MDR extension followed in March 2023. This extension not only helps manufacturers but also provides notified bodies with more time to process certifications, which is crucial for allowing devices to continue being available on the market.
MDR Transitional Periods
The extended transitional periods under the MDR vary depending on the risk class of the device:
- Class III and Class IIb Implantable Devices: These can remain on the market until December 31, 2027. Devices based on well-established technologies, for which the MDR provides specific exemptions, are not included in this extension.
- Other Risk Classes: Devices from other risk classes have until December 31, 2028, to comply.
- Custom-Made Class III Devices: These can be marketed or put into service without a certificate from a notified body until May 26, 2026.
- Devices Requiring Notified Body Involvement Post-MDR: Devices that did not require a notified body under the MDD but do under the MDR, and had a declaration of conformity drawn up before May 26, 2021, can be marketed or put into service until December 31, 2028.
- The use of these extended periods is contingent upon the devices meeting the safety conditions outlined in regulation 2023/607.
IVDR Transitional Periods
For IVDs, the transitional periods are set based on the device's risk class:
- Class D Devices: Must comply by May 26, 2025.
- Class C Devices: Have until May 26, 2026.
- Class B and Class A Devices (Sterile): Need to meet the requirements by May 26, 2027.
- The Health and Youth Care Inspectorate (IGJ) urges manufacturers to comply with the IVDR requirements ahead of these deadlines to ensure device safety and effectiveness.
These transitional provisions are designed to balance the need for regulatory compliance with the practicalities of manufacturing and certifying a wide array of medical devices and IVDs. Manufacturers are encouraged to use this period to fully align their products with the new regulatory standards to ensure continued market access and patient safety.
Restrictions on Substances under REACH & CLP:
Similar to the EU MDR, the EU IVDR also imposes restrictions on the use of substances under the REACH and CLP regulations. Manufacturers of IVD devices must comply with these regulations by identifying and managing substances of very high concern (SVHC) present in their devices. If an SVHC exceeds 0.1% weight by weight, manufacturers must provide information to downstream users and consumers, including safe handling instructions and potential risks associated with the substance. Additionally, the CLP regulation provides harmonized criteria for classifying, labeling, and packaging chemicals. Medical device manufacturers must comply with CLP requirements by correctly classifying hazardous substances present in their devices and providing appropriate labels and safety data sheets to downstream users.
Conclusion: Compliance with the EU MDR and EU IVDR is crucial for medical device manufacturers operating in the European Union. These regulations ensure patient safety, harmonize standards and promote the highest level of quality and performance for medical devices. Manufacturers must diligently adhere to the compliance requirements, including classification, Technical Documentation, involvement with Notified Bodies, and restrictions on substances under REACH and CLP. By doing so, manufacturers can navigate the regulatory landscape, gain market access, and contribute to the advancement of healthcare in the EU.
To navigate the complex landscape of medical device regulations in the EU, manufacturers may seek guidance from industry experts, consultants, or regulatory affairs professionals specializing in EU MDR and EU IVDR compliance. Book a demo today to ensure a smooth and successful compliance journey for your medical devices.
 Is a Smart Business Investment — Even If You’re Not in Compliance")

