Table of Contents
La industria de dispositivos médicos desempeña un papel crucial en el avance de la atención médica a nivel mundial, y garantizar la seguridad del paciente y la eficacia del producto es de suma importancia. En la Unión Europea (UE), los fabricantes de dispositivos médicos deben cumplir con regulaciones específicas para asegurar que sus productos cumplan con los más altos estándares. Las dos regulaciones clave que rigen los dispositivos médicos en la UE son el Reglamento de Dispositivos Médicos de la UE (EU MDR) y el Reglamento de Diagnóstico In Vitro de la UE (EU IVDR). En este artículo, profundizaremos en los requisitos de cumplimiento detallados por estas regulaciones y sus implicaciones para los fabricantes.
¿Qué es el Reglamento de Dispositivos Médicos EU MDR?
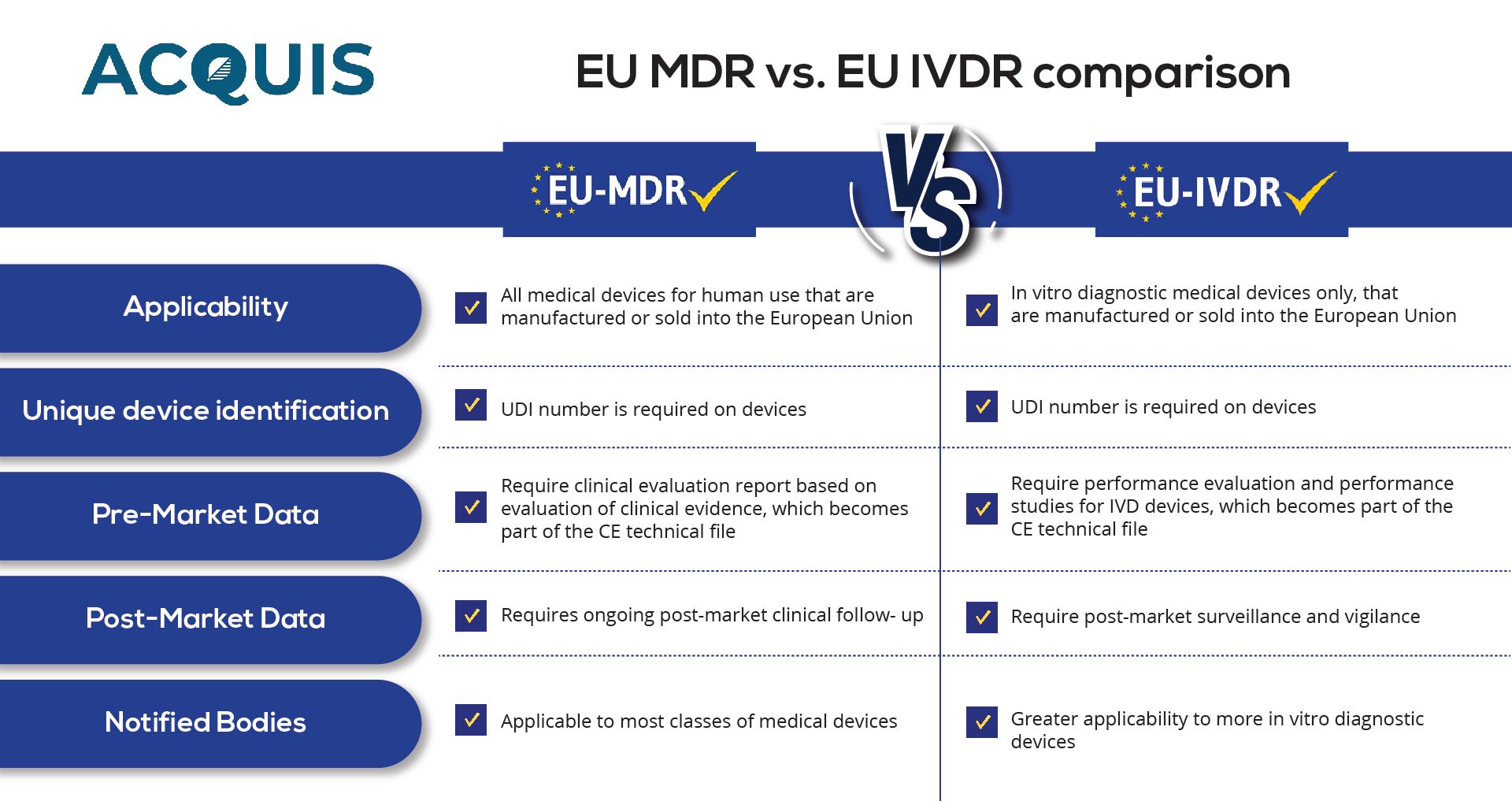
El Reglamento de Dispositivos Médicos (EU MDR), efectivo desde el 26 de mayo de 2021, representa una revisión significativa de las directivas anteriores. Este reglamento se estableció para reemplazar la Directiva de Dispositivos Médicos (93/42/EEC) y la Directiva para Dispositivos Médicos Implantables Activos (90/385/EEC). El MDR introduce requisitos más estrictos para los fabricantes de dispositivos médicos, enfocándose en una evaluación clínica rigurosa y vigilancia del mercado para mejorar la calidad y seguridad de los dispositivos. El alcance ampliado del reglamento cubre más tipos de dispositivos, aumentando la responsabilidad de los fabricantes y otras partes interesadas en la cadena de suministro.
Cambios Clave: Uno de los cambios más notables bajo el MDR es el requisito de un sistema integral de gestión de calidad y la participación obligatoria de un representante autorizado europeo para fabricantes no pertenecientes a la UE.
Impacto: El MDR busca reducir los riesgos asociados con los dispositivos médicos en el mercado mediante la mejora de la transparencia y trazabilidad a lo largo del ciclo de vida del dispositivo.
El amplio alcance de la Sección 10.4.1 del EU MDR:
La Sección 10.4.1 del Reglamento de Dispositivos Médicos de la UE (EU MDR) desempeña un papel crucial en garantizar la seguridad y calidad de los dispositivos médicos. Esta sección requiere que los fabricantes identifiquen ciertas sustancias en sus productos. Específicamente, los fabricantes deben identificar carcinógenos, mutágenos, toxinas reproductivas (CMRs) y químicos que alteran el sistema endocrino (EDCs) presentes en dispositivos invasivos o materiales que entren en contacto con fluidos o gases. Estas sustancias deben identificarse si están presentes en concentraciones que superan el 0.1% p/p.
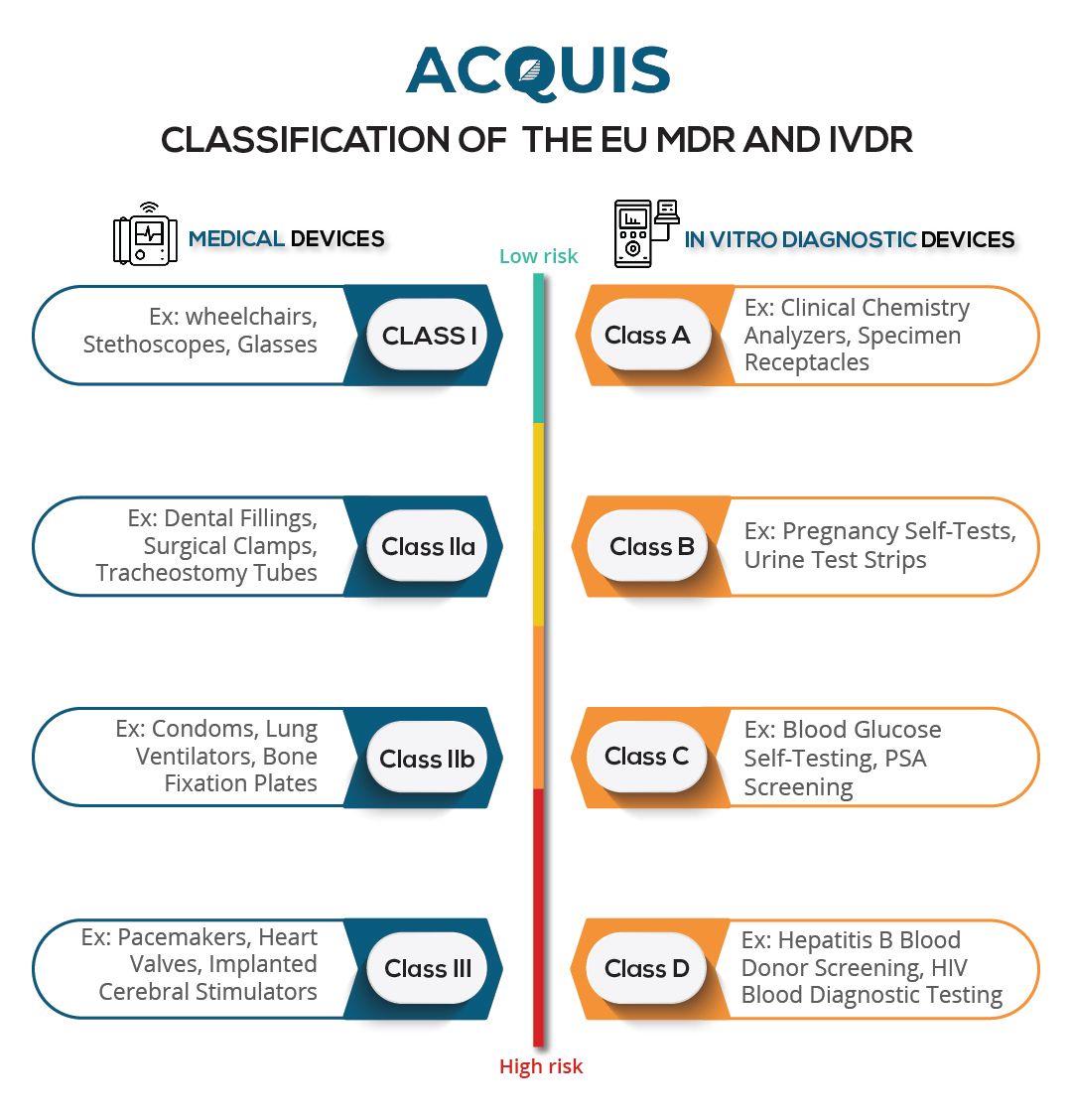
Bajo el EU MDR, los fabricantes de dispositivos médicos deben adherirse a varios requisitos clave de cumplimiento. En primer lugar, se requiere que los fabricantes clasifiquen sus dispositivos según el riesgo y los asignen a una de las cuatro clases: Clase I, Clase IIa, Clase IIb, o Clase III. Esta clasificación determina el nivel de escrutinio y los procedimientos de evaluación de la conformidad necesarios.
El proceso de clasificación sigue 22 reglas descritas en el Anexo VIII del EU MDR. Las reglas 19–22 son nuevas en el EU MDR, mientras que las reglas 1–18 han sido trasladadas de la MDD anterior. Al considerar los tres criterios especificados en la Sección 10.4.1, en conjunto con estas reglas de clasificación, las empresas pueden determinar el impacto del EU MDR en sus productos.
 Los fabricantes también deben compilar y mantener una Documentación Técnica completa, que incluya información detallada sobre el diseño, fabricación y rendimiento del dispositivo. Esta documentación debe demostrar el cumplimiento con los requisitos generales de seguridad y rendimiento establecidos en la regulación. También debe incluir un Informe de Evaluación Clínica (CER) basado en datos clínicos recopilados para respaldar las afirmaciones de seguridad y rendimiento del dispositivo.
Los fabricantes también deben compilar y mantener una Documentación Técnica completa, que incluya información detallada sobre el diseño, fabricación y rendimiento del dispositivo. Esta documentación debe demostrar el cumplimiento con los requisitos generales de seguridad y rendimiento establecidos en la regulación. También debe incluir un Informe de Evaluación Clínica (CER) basado en datos clínicos recopilados para respaldar las afirmaciones de seguridad y rendimiento del dispositivo.
Otro aspecto importante del cumplimiento con el EU MDR es el requisito de un Organismo Notificado. Los Organismos Notificados son organizaciones independientes designadas por los estados miembros de la UE para evaluar la conformidad de los dispositivos médicos. Los fabricantes deben obtener un certificado válido de un Organismo Notificado para demostrar el cumplimiento con la regulación. El proceso de certificación implica una evaluación exhaustiva de la Documentación Técnica, el Sistema de Gestión de la Calidad y, en algunos casos, una revisión de la evidencia clínica.
¿Qué es el Reglamento de Diagnóstico In Vitro EU IVDR?
El Reglamento de Diagnóstico In Vitro (EU IVDR) entró en vigor el 26 de mayo de 2022, reemplazando la antigua Directiva de la UE sobre dispositivos médicos de diagnóstico in vitro (98/79/CE). Este reglamento establece nuevos estándares para la aprobación y vigilancia de los IVD, que son herramientas utilizadas para realizar pruebas en muestras del cuerpo humano. El IVDR introduce un nuevo sistema de clasificación basado en el riesgo asociado con el dispositivo, requiriendo documentación y cumplimiento más estrictos para las clases de mayor riesgo.
Bajo el EU IVDR, los fabricantes de IVD enfrentan requisitos de cumplimiento estrictos. Un cambio significativo es la introducción de un sistema de clasificación basado en el riesgo para los dispositivos IVD.
El reglamento incluye cuatro clases: Clase A, Clase B, Clase C y Clase D. Los dispositivos de mayor riesgo requerirán la participación de un Organismo Notificado para evaluar la conformidad.
Para cumplir con el EU IVDR, los fabricantes deben establecer un Sistema de Gestión de la Calidad (QMS) y realizar una Documentación Técnica completa. La Documentación Técnica debe incluir información detallada sobre el diseño del dispositivo, el propósito previsto, el rendimiento analítico y clínico, así como una evaluación de cualquier afirmación realizada. La evidencia clínica, como los estudios de evaluación de rendimiento o los estudios de rendimiento clínico, debe respaldar las afirmaciones del dispositivo.
Al igual que el EU MDR, el EU IVDR también requiere la participación de un Organismo Notificado. Los fabricantes deben obtener un certificado válido de un Organismo Notificado para demostrar el cumplimiento con la regulación. Los Organismos Notificados revisarán la Documentación Técnica y evaluarán la conformidad del dispositivo con los requisitos del reglamento.
Disposiciones Transitorias para Dispositivos Médicos y IVD
La implementación del MDR y el IVDR ha introducido disposiciones transitorias que permiten que ciertos dispositivos médicos y IVD, denominados "dispositivos heredados", continúen comercializándose bajo las directivas anteriores por un tiempo limitado. Estas disposiciones aseguran que los fabricantes tengan tiempo suficiente para cumplir con los nuevos requisitos regulatorios y obtener la necesaria certificación CE.
Dispositivos Heredados y Plazos ExtendidosLos dispositivos heredados son aquellos que fueron certificados bajo las antiguas directivas antes de que se aplicaran las nuevas regulaciones. Para prevenir interrupciones en el mercado y riesgos para la seguridad del paciente, se han extendido los plazos para el cumplimiento tanto del MDR como del IVDR. La IVDR se extendió en enero de 2022, mientras que la extensión del MDR siguió en marzo de 2023. Esta extensión no solo ayuda a los fabricantes, sino que también proporciona a los organismos notificados más tiempo para procesar certificaciones, lo cual es crucial para permitir que los dispositivos continúen estando disponibles en el mercado.
Períodos de Transición del MDR
Los períodos de transición extendidos bajo el MDR varían dependiendo de la clase de riesgo del dispositivo:
- Dispositivos Implantables de Clase III y Clase IIb: Estos pueden permanecer en el mercado hasta el 31 de diciembre de 2027. Los dispositivos basados en tecnologías bien establecidas, para los cuales el MDR proporciona exenciones específicas, no están incluidos en esta extensión.
- Otras Clases de Riesgo: Los dispositivos de otras clases de riesgo tienen hasta el 31 de diciembre de 2028 para cumplir.
- Dispositivos a Medida de Clase III: Estos pueden ser comercializados o puestos en servicio sin un certificado de un organismo notificado hasta el 26 de mayo de 2026.
- Dispositivos que Requieren Involucramiento de un Organismo Notificado Post-MDR: Dispositivos que no requerían un organismo notificado bajo el MDD pero lo hacen bajo el MDR, y tenían una declaración de conformidad elaborada antes del 26 de mayo de 2021, pueden ser comercializados o puestos en servicio hasta el 31 de diciembre de 2028.
- El uso de estos períodos extendidos depende de que los dispositivos cumplan con las condiciones de seguridad descritas en la regulación 2023/607.
Períodos de Transición del IVDR
Para los IVD, los períodos de transición se establecen en función de la clase de riesgo del dispositivo:
- Dispositivos de Clase D: Deben cumplir antes del 26 de mayo de 2025.
- Dispositivos de Clase C: Tienen hasta el 26 de mayo de 2026.
- Dispositivos de Clase B y Clase A (Esterilizados): Necesitan cumplir con los requisitos antes del 26 de mayo de 2027.
- La Inspección de Salud y Cuidado Juvenil (IGJ) insta a los fabricantes a cumplir con los requisitos del IVDR antes de estas fechas para garantizar la seguridad y eficacia de los dispositivos.
Estas disposiciones transitorias están diseñadas para equilibrar la necesidad de cumplimiento regulatorio con las realidades de la fabricación y certificación de una amplia variedad de dispositivos médicos e IVD. Se anima a los fabricantes a utilizar este período para alinear completamente sus productos con los nuevos estándares regulatorios para asegurar el acceso continuado al mercado y la seguridad del paciente.
Restricciones sobre Sustancias bajo REACH & CLP:
Similar al EU MDR, el EU IVDR también impone restricciones sobre el uso de sustancias bajo las regulaciones REACH y CLP. Los fabricantes de dispositivos IVD deben cumplir con estas regulaciones identificando y gestionando las sustancias extremadamente preocupantes (SVHC) presentes en sus dispositivos. Si una SVHC excede el 0.1% por peso, los fabricantes deben proporcionar información a los usuarios finales y consumidores, incluyendo instrucciones de manejo seguro y posibles riesgos asociados con la sustancia. Adicionalmente, la regulación CLP proporciona criterios armonizados para la clasificación, etiquetado y embalaje de productos químicos. Los fabricantes de dispositivos médicos deben cumplir con los requisitos del CLP clasificando correctamente las sustancias peligrosas presentes en sus dispositivos y proporcionando las etiquetas y hojas de datos de seguridad apropiadas a los usuarios finales.Conclusión: Cumplir con el EU MDR y el EU IVDR es crucial para los fabricantes de dispositivos médicos que operan en la Unión Europea. Estas normativas aseguran la seguridad del paciente, armonizan los estándares y promueven el más alto nivel de calidad y rendimiento para los dispositivos médicos. Los fabricantes deben adherirse diligentemente a los requisitos de cumplimiento, incluyendo la clasificación, la Documentación Técnica, la participación de Organismos Notificados y las restricciones sobre sustancias bajo REACH y CLP. Al hacerlo, los fabricantes pueden navegar por el panorama regulatorio, acceder al mercado y contribuir al avance de la atención sanitaria en la UE.
Para navegar por el complejo panorama de las regulaciones de dispositivos médicos en la UE, los fabricantes pueden buscar orientación de expertos de la industria, consultores o profesionales de asuntos regulatorios especializados en cumplimiento de EU MDR y EU IVDR. Reserve una demostración hoy para asegurar un viaje de cumplimiento exitoso y sin problemas para sus dispositivos médicos.