Table of Contents
Die Medizingeräteindustrie spielt eine entscheidende Rolle bei der Weiterentwicklung der Gesundheitsversorgung weltweit, und die Gewährleistung der Patientensicherheit und der Produktwirksamkeit hat höchste Priorität. In der Europäischen Union (EU) müssen Hersteller von Medizinprodukten spezifische Vorschriften einhalten, um sicherzustellen, dass ihre Produkte den höchsten Standards entsprechen. Die beiden wichtigsten Vorschriften, die Medizinprodukte in der EU regeln, sind die EU-Verordnung über Medizinprodukte (EU-MDR) und die EU-Verordnung über In-vitro-Diagnostika (EU-IVDR). In diesem Artikel werden wir die in diesen Verordnungen festgelegten Compliance-Anforderungen und deren Auswirkungen auf Hersteller erörtern.
Was ist die EU-MDR-Verordnung über Medizinprodukte:
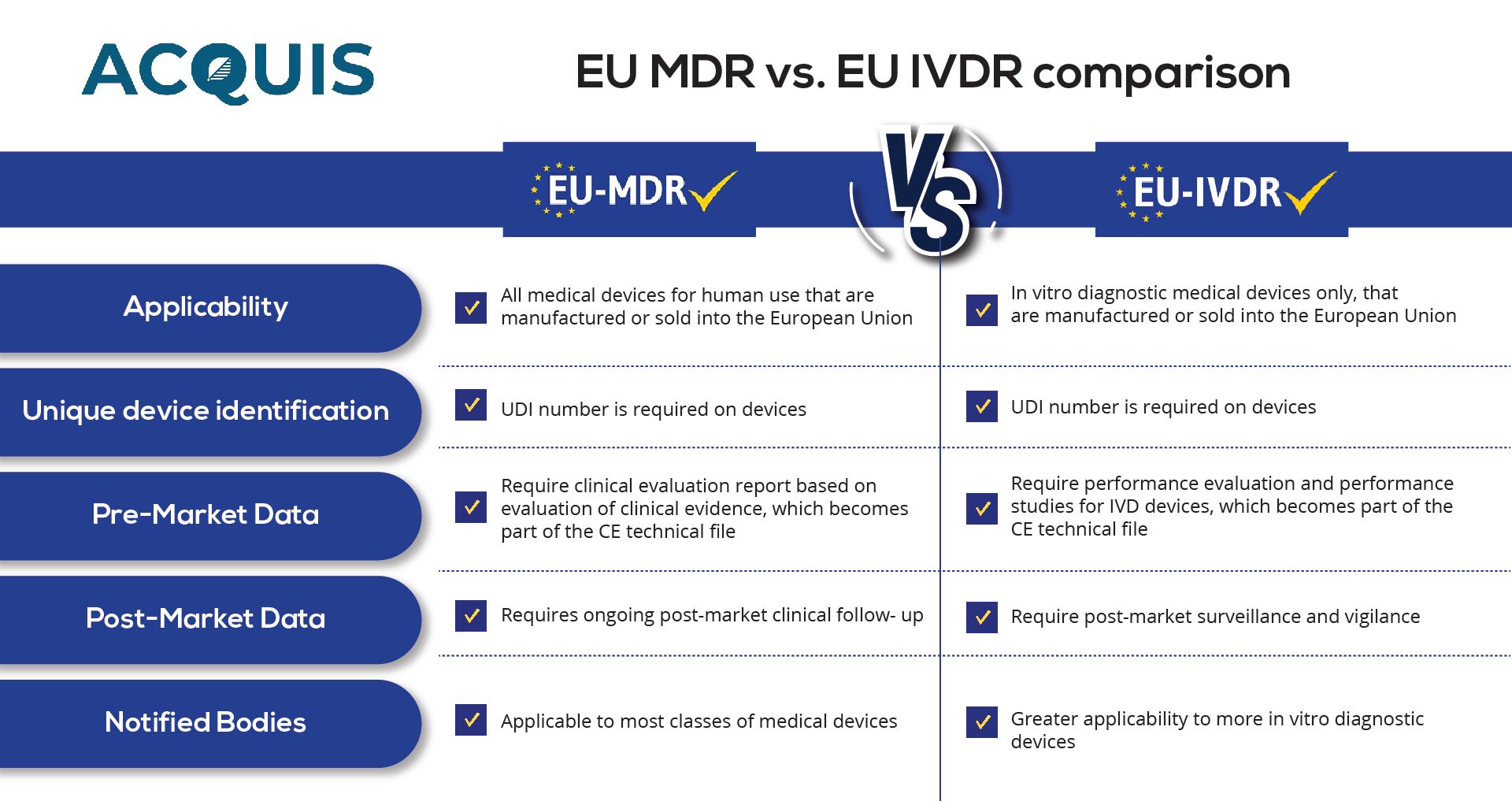
Die Verordnung über Medizinprodukte (EU-MDR), die seit dem 26. Mai 2021 in Kraft ist, stellt eine bedeutende Überarbeitung der vorherigen Richtlinien dar. Diese Verordnung wurde eingeführt, um die Richtlinie über Medizinprodukte (93/42/EWG) und die Richtlinie für aktive implantierbare medizinische Geräte (90/385/EWG) zu ersetzen. Die MDR führt erweiterte Anforderungen für Hersteller von Medizinprodukten ein und konzentriert sich auf eine umfassende klinische Bewertung und Marktüberwachung zur Verbesserung der Qualität und Sicherheit von Geräten. Der erweiterte Geltungsbereich der Verordnung umfasst mehr Gerätetypen und erhöht die Verantwortlichkeit der Hersteller und anderer Stakeholder in der Versorgungskette.
Wesentliche Änderungen: Eine der bemerkenswertesten Änderungen durch die MDR ist die Anforderung eines umfassenden Qualitätsmanagementsystems und die obligatorische Beteiligung eines in Europa zugelassenen Vertreters für Hersteller außerhalb der EU.
Auswirkung: Die MDR zielt darauf ab, die mit medizinischen Geräten verbundenen Risiken auf dem Markt zu reduzieren, indem sie die Transparenz und Rückverfolgbarkeit im gesamten Lebenszyklus des Geräts erhöht.
Der breite Geltungsbereich des Abschnitts 10.4.1 der EU-MDR:
Abschnitt 10.4.1 der EU-Verordnung über Medizinprodukte (EU-MDR) spielt eine entscheidende Rolle bei der Gewährleistung der Sicherheit und Qualität von Medizinprodukten. Dieser Abschnitt verlangt von den Herstellern die Identifizierung bestimmter Substanzen in ihren Produkten. Insbesondere müssen Hersteller krebserregende, erbgutverändernde und reproduktionsgefährdende Stoffe (CMR) sowie endokrin wirksame Chemikalien (EDC) identifizieren, die in invasiven Geräten oder Materialien enthalten sind, die mit Flüssigkeiten oder Gasen in Kontakt kommen. Diese Substanzen müssen identifiziert werden, wenn sie in Konzentrationen von mehr als 0,1% w/w vorliegen.
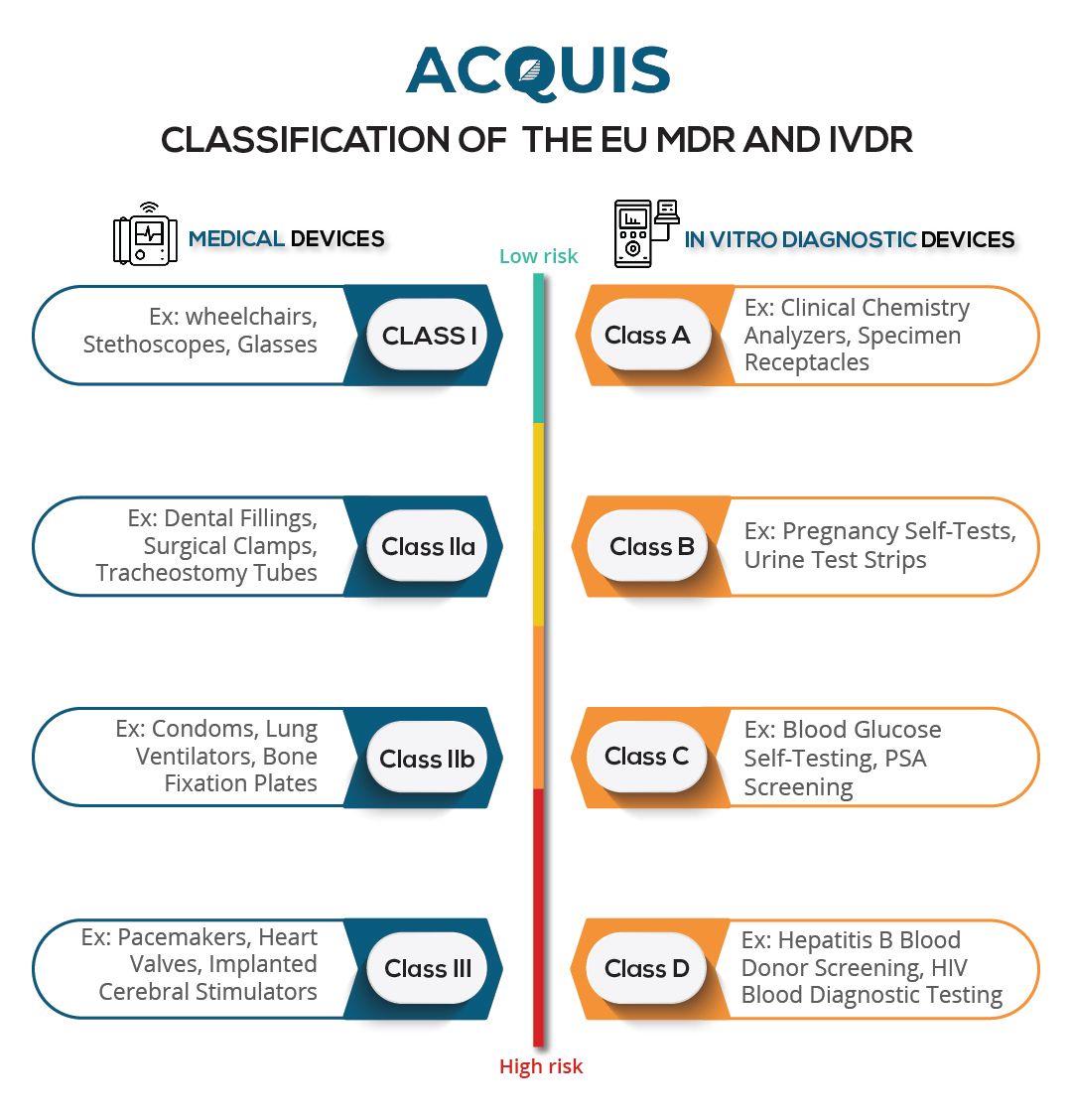
Hersteller von Medizinprodukten müssen gemäß der EU-MDR mehreren wichtigen Compliance-Anforderungen nachkommen. Zunächst müssen Hersteller ihre Produkte nach Risiko klassifizieren und einer von vier Klassen zuweisen: Klasse I, Klasse IIa, Klasse IIb oder Klasse III. Diese Klassifizierung bestimmt das Maß an Kontrolle und die notwendigen Konformitätsbewertungsverfahren.
Der Klassifizierungsprozess folgt 22 Regeln, die in EU-MDR Anhang VIII festgelegt sind. Die Regeln 19–22 sind neu in der EU-MDR, während die Regeln 1–18 aus der vorherigen MDD übernommen wurden. Indem Unternehmen die drei in Abschnitt 10.4.1 angegebenen Kriterien in Verbindung mit diesen Klassifizierungsregeln berücksichtigen, können sie die Auswirkungen der EU-MDR auf ihre Produkte bestimmen.
 Hersteller müssen außerdem umfassende Technische Dokumentationen erstellen und pflegen, die detaillierte Informationen über das Design, die Herstellung und die Leistung des Geräts enthalten. Diese Dokumentation sollte die Einhaltung der allgemeinen Sicherheits- und Leistungsanforderungen belegen, die in der Verordnung festgelegt sind. Sie sollte auch einen Klinischen Bewertungsbericht (Clinical Evaluation Report, CER) enthalten, der auf klinischen Daten basiert, die zur Unterstützung der Sicherheits- und Leistungsaussagen des Geräts gesammelt wurden.
Hersteller müssen außerdem umfassende Technische Dokumentationen erstellen und pflegen, die detaillierte Informationen über das Design, die Herstellung und die Leistung des Geräts enthalten. Diese Dokumentation sollte die Einhaltung der allgemeinen Sicherheits- und Leistungsanforderungen belegen, die in der Verordnung festgelegt sind. Sie sollte auch einen Klinischen Bewertungsbericht (Clinical Evaluation Report, CER) enthalten, der auf klinischen Daten basiert, die zur Unterstützung der Sicherheits- und Leistungsaussagen des Geräts gesammelt wurden.
Ein weiterer wichtiger Aspekt der EU MDR-Konformität ist die Anforderung eines Benannten Stelle. Benannte Stellen sind unabhängige Organisationen, die von den EU-Mitgliedsstaaten benannt werden, um die Konformität von Medizinprodukten zu bewerten. Hersteller müssen ein gültiges Zertifikat von einer Benannten Stelle erhalten, um die Einhaltung der Verordnung nachzuweisen. Der Zertifizierungsprozess umfasst eine gründliche Bewertung der Technischen Dokumentation, des Qualitätsmanagementsystems und in einigen Fällen eine Überprüfung klinischer Nachweise.
Was ist die EU-IVDR-Verordnung über In-vitro-Diagnostika
Die Verordnung über In-vitro-Diagnostika (EU IVDR) trat am 26. Mai 2022 in Kraft und ersetzt die älteren EU-Richtlinien über In-vitro-Diagnostika (98/79/EG). Diese Verordnung legt neue Standards für die Zulassung und Überwachung von IVDs, die zur Durchführung von Tests an Proben des menschlichen Körpers verwendet werden, fest. Die IVDR führt ein neues Klassifizierungssystem basierend auf dem mit dem Gerät verbundenen Risiko ein, das für höherklassifizierte Risikoklassen strengere Dokumentations- und Konformitätsanforderungen erfordert.
Unter der EU IVDR stehen IVD-Hersteller vor strengen Konformitätsanforderungen. Eine wesentliche Änderung ist die Einführung eines risikobasierten Klassifizierungssystems für IVD-Geräte.
Die Verordnung umfasst vier Klassen: Klasse A, Klasse B, Klasse C und Klasse D. Geräte mit höherem Risiko erfordern die Beteiligung einer Benannten Stelle zur Bewertung der Konformität.
Um die EU IVDR einzuhalten, müssen Hersteller ein Qualitätsmanagementsystem (QMS) einrichten und umfassende Technische Dokumentationen durchführen. Die Technische Dokumentation muss detaillierte Informationen über das Design des Geräts, den beabsichtigten Zweck, die analytische und klinische Leistung sowie eine Bewertung der geltend gemachten Aussagen enthalten. Klinische Nachweise, wie Leistungsbewertungsstudien oder klinische Leistungsstudien, müssen die Aussagen des Geräts unterstützen.
Wie bei der EU MDR erfordert auch die EU IVDR die Beteiligung einer Benannten Stelle. Hersteller müssen ein gültiges Zertifikat von einer Benannten Stelle erhalten, um die Konformität mit der Verordnung nachzuweisen. Benannte Stellen überprüfen die Technische Dokumentation und bewerten die Konformität des Geräts mit den Anforderungen der Verordnung.
Übergangsbestimmungen für Medizinprodukte und IVDs
Die Umsetzung der MDR und IVDR hat Übergangsbestimmungen eingeführt, die es bestimmten Medizinprodukten und IVDs, den sogenannten „Legacy Devices“, ermöglichen, für einen begrenzten Zeitraum weiterhin unter den vorherigen Richtlinien vermarktet zu werden. Diese Bestimmungen stellen sicher, dass Hersteller ausreichende Zeit haben, um die neuen regulatorischen Anforderungen zu erfüllen und die erforderliche CE-Zertifizierung zu erhalten.
Legacy Devices und verlängerte FristenLegacy-Geräte sind solche, die nach den alten Richtlinien zertifiziert wurden, bevor die neuen Vorschriften in Kraft traten. Um Marktstörungen und Risiken für die Patientensicherheit zu vermeiden, wurden die Fristen für die Einhaltung sowohl der MDR als auch der IVDR verlängert. Die IVDR wurde im Januar 2022 verlängert, während die MDR-Verlängerung im März 2023 folgte. Diese Verlängerung hilft nicht nur den Herstellern, sondern gibt auch den benannten Stellen mehr Zeit, Zertifizierungen zu bearbeiten, was entscheidend dafür ist, dass Geräte weiterhin auf dem Markt verfügbar bleiben.
MDR-Übergangszeiträume
Die verlängerten Übergangszeiträume unter der MDR variieren je nach Risikoklasse des Geräts:
- Klasse III und Klasse IIb Implantierbare Geräte: Diese können bis zum 31. Dezember 2027 auf dem Markt bleiben. Geräte auf Basis gut etablierter Technologien, für die die MDR spezifische Ausnahmen vorsieht, sind nicht in dieser Verlängerung eingeschlossen.
- Andere Risikoklassen: Geräte aus anderen Risikoklassen haben bis zum 31. Dezember 2028 Zeit zur Einhaltung.
- Sonderanfertigungen der Klasse III: Diese können ohne Zertifikat einer benannten Stelle bis zum 26. Mai 2026 vermarktet oder in Betrieb genommen werden.
- Geräte mit Benannte-Stelle-Beteiligung nach MDR: Geräte, die unter der MDD keine benannte Stelle benötigten, aber unter der MDR, und eine Konformitätserklärung vor dem 26. Mai 2021 erstellt wurde, können bis zum 31. Dezember 2028 vermarktet oder in Betrieb genommen werden.
- Die Nutzung dieser verlängerten Fristen hängt davon ab, dass die Geräte die in der Verordnung 2023/607 beschriebenen Sicherheitsbedingungen erfüllen.
IVDR-Übergangszeiträume
Für IVDs sind die Übergangsfristen basierend auf der Risikoklasse des Geräts festgelegt:
- Geräte der Klasse D: Müssen bis zum 26. Mai 2025 konform sein.
- Geräte der Klasse C: Haben bis zum 26. Mai 2026 Zeit.
- Geräte der Klasse B und Klasse A (Steril): Müssen die Anforderungen bis zum 26. Mai 2027 erfüllen.
- Die Gesundheits- und Jugendpflegeaufsicht (IGJ) drängt die Hersteller, die IVDR-Anforderungen vor diesen Terminen zu erfüllen, um die Sicherheit und Wirksamkeit der Geräte zu gewährleisten.
Diese Übergangsbestimmungen zielen darauf ab, den Bedarf an regulatorischer Einhaltung mit den praktischen Anforderungen der Herstellung und Zertifizierung eines breiten Spektrums von Medizinprodukten und IVDs in Einklang zu bringen. Die Hersteller werden ermutigt, diesen Zeitraum zu nutzen, um ihre Produkte vollständig mit den neuen regulatorischen Standards in Einklang zu bringen, um einen kontinuierlichen Marktzugang und Patientensicherheit zu gewährleisten.
Beschränkungen für Stoffe gemäß REACH & CLP:
Ähnlich wie die EU-MDR legt auch die EU-IVDR Beschränkungen für die Verwendung von Stoffen gemäß den REACH- und CLP-Verordnungen fest. Hersteller von IVD-Geräten müssen diese Vorschriften einhalten, indem sie Stoffe von sehr hoher Besorgnis (SVHC) in ihren Geräten identifizieren und verwalten. Wenn ein SVHC 0,1% Gewichtsprozent überschreitet, müssen Hersteller Informationen an nachgeschaltete Anwender und Verbraucher bereitstellen, einschließlich sicherer Handhabungsanweisungen und potenzieller Risiken im Zusammenhang mit der Substanz. Darüber hinaus bietet die CLP-Verordnung harmonisierte Kriterien für die Einstufung, Kennzeichnung und Verpackung von Chemikalien. Medizinproduktehersteller müssen die CLP-Anforderungen einhalten, indem sie gefährliche Stoffe in ihren Geräten korrekt einstufen und geeignete Etiketten und Sicherheitsdatenblätter an nachgeschaltete Anwender bereitstellen.Schlussfolgerung: Die Einhaltung der EU-MDR und EU-IVDR ist entscheidend für Hersteller von Medizinprodukten, die in der Europäischen Union tätig sind. Diese Vorschriften gewährleisten die Patientensicherheit, harmonisieren Standards und fördern das höchste Maß an Qualität und Leistung bei Medizinprodukten. Hersteller müssen die Anforderungen an die Konformität sorgfältig einhalten, einschließlich Klassifizierung, Technische Dokumentation, Einbindung Benannter Stellen und Beschränkungen für Stoffe gemäß REACH und CLP. Auf diese Weise können Hersteller sich im regulatorischen Umfeld zurechtfinden, Marktzugang erlangen und zur Weiterentwicklung des Gesundheitswesens in der EU beitragen.
Um sich im komplexen Umfeld der Vorschriften für Medizinprodukte in der EU zurechtzufinden, können Hersteller Rat bei Industrieexperten, Beratern oder Fachleuten für Regulierungsfragen einholen, die auf die Einhaltung von EU-MDR und EU-IVDR spezialisiert sind. Buchen Sie eine Demo, um eine reibungslose und erfolgreiche Konformitätsreise für Ihre Medizinprodukte sicherzustellen.